Dr. Jan Mitschker (Former PhD Student)
Dr. Jan Mitschker (Former PhD Student)
Research interests

Photocatalytic water splitting on titanium dioxide
- ab initio quantum chemistry
- quantum dynamics
- artificial neural networks for fitting of potential energy surface
See also Priority Program 1613
Dissertation (2015)
Title:
Quantum chemical and quantum dynamical investigation on the photochemistry of water on titanium dioxide surfaces
A pdf-version of this dissertation is available on the servers of the university's library verfügbar.
Abstract (German)
In this dissertation, the photochemistry of water on rutile is investigated.
The surface photochemistry of water is important for a number of applications and could represent a new approach to hydrogen.
A cluster embedded in a point charge field serves as a model for the surface.
Systematic investigations ensured that parameters such as cluster size, basis set and method provide a sufficiently good description of the system.
It was shown that a Ti₉O₁₈Mg₇14+ cluster, as recently developed for CO photodesorption, can also be used for water adsorption and dissociation.
Both molecular and dissociative adsorption could be identified on this cluster, with the former being the more stable.
By analysing their geometries, five spatial degrees of freedom were identified as important for the potential surfaces.
The construction of these surfaces was enormously complicated by the multireference character of the wave function that occurs during dissociation.
Therefore, several CASSCF approaches with different active spaces were investigated until finally a solution strategy was developed by freezing inactive orbitals.
The topology of the potential surface of the electronic ground state is very complex, which is caused by the different minima and the coupling of all degrees of freedom. The surface was constructed from more than 240000 data points and fitted with an artificial neural network. This approach was very successful and only led to small errors.
The electronically excited state was realised by removing an electron from the water molecule. This approach is consistent with the experimental observation of holes attacking the adsorbate. In molecular adsorption, the state is dominated by strong repulsive forces between the adsorbate and the surface. Dissociative adsorption, on the other hand, is further favoured.
The potential surfaces were used for quantum dynamic simulations in which the adsorbate is described as a wave packet. The photodesorption of water was studied using the jumping wave packet approach. It was shown that this reaction is an example of the MGR mechanism and leads to rapidly desorbing molecules.
The photodissociation of water was investigated as a second reaction. The OH group was fixed and only the movement of a hydrogen atom was simulated. The dynamics are determined by a small energy barrier near the Franck-Condon point. This reduces the probability of dissociation and leads to pronounced isotope effects.
Abstract (English)
In this thesis, the photochemistry of water on rutile is studied from first principles.
Surface photochemistry of water is important for a variety of applications and may lead to new routes for hydrogen production.
A cluster embedded in a finite point charge field is used as a model surface.
Systematic investigations ensured that parameters like cluster size, basis set and methods are sufficient for a good description of the water/rutile interaction.
It could be shown that a Ti9O18Mg714+ cluster, recently used for CO photodesorption, is large enough for water adsorption and dissociation, too.
On this cluster, a molecular and a dissociative adsorption form was found, the former being the most stable one.
Analyzing their geometries, five spatial degrees of freedom were identified as important for potential energy surfaces.
Construction of these surfaces is complicated due to the multi-reference character of the wave function in the course of the dissociation process.
Therefore, within the CASSCF approach a variety of active spaces was investigated. Finally, a two-step solution freezing the inactive orbitals paved the way for the potential energy surface.
The potential energy surface of the electronic ground state is very complicated because of different minima and a strong coupling between all degrees of freedom. The surface was constructed from more than 240000 data points and fitted by means of an artificial neural network. This approach proved to be very reliable giving only small errors and a good interpolation.
For the electronically excited state, one electron was removed from the water molecule. This is justified by the experimental observation of a hole attacking the adsorbate. This state is dominated by strong repulsive interactions between the adsorbate and the surface, as far as the molecular adsorption form is concerned. However, the dissociative form becomes even more favourite.
The potential energy surfaces were used for quantum dynamical simulations describing the adsorbate as a wave packet. The photodesorption of water in three dimensions was calculated within the jumping wave packet approach. The reaction was found to be a prototype of the MGR mechanism with very fast desorbing molecules.
The second reaction studied in this thesis is the photodissociation of water. Here, a simple system with a fixed \OH group was used to study the motion of hydrogen. The dynamics is controlled by a small energy barrier near the Franck--Condon point. This decreases the dissociation probability and leads to pronounced isotope effects.
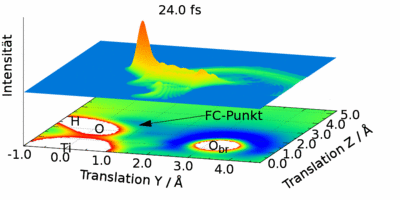
Quantum dynamic description of a hydrogen atom as a wave packet.
The temporal development of the magnitude squared wave function for a fixed OH group on the rutile surface is shown. The positions of some atoms and the potential are also shown.