Dr. Jan Mitschker (Ehemaliger Doktorand)
Kontakt
Dr. Jan Mitschker (Ehemaliger Doktorand)
Forschungsgebiet

Photokatalytische Wasserspaltung auf Titandioxid
- ab initio Quantenchemie
- Quantendynamik
- Neuronale Netze zum Fitten von Potentialflächen
Siehe auch Schwerpunktprogramm 1613
Dissertation (2015)
Titel:
Quantenchemische und quantendynamische Untersuchungen zur Photochemie von Wasser auf einer Titandioxidoberfläche
Eine pdf-Version der Dissertation ist auf dem Server der Universitätsbibliothek verfügbar.
Kurzfassung (Deutsch)
In dieser Dissertation wird die Photochemie von Wasser auf Rutil untersucht.
Die Oberflächenphotochemie von Wasser ist für eine Reihe von Anwendungen wichtig und könnte einen neuen Zugang zu Wasserstoff darstellen.
Ein in ein Punktladungsfeld eingebetteter Cluster dient als Modell für die Oberfläche.
Systematische Untersuchungen stellten sicher, dass Parameter wie Clustergröße, Basissatz und Methode eine hinreichend gute Beschreibung des Systems liefern.
Es konnte gezeigt werden, dass ein Ti9O18Mg714+-Cluster, wie er kürzlich für die CO-Photodesorption entwickelt worden ist, auch für die Wasseradsorption und -dissoziation genutzt werden kann.
Auf diesem Cluster konnte sowohl eine molekulare als auch eine dissoziative Adsorption ausgemacht werden, wobei erstere die stabilere ist.
Durch Analyse ihrer Geometrien konnten fünf räumliche Freiheitsgrade als für die Potentialflächen wichtig identifiziert werden.
Die Konstruktion dieser Flächen wurde durch den Multireferenzcharakter der Wellenfunktion, der im Verlauf der Dissoziation auftritt, enorm erschwert.
Daher wurden mehrere CASSCF-Ansätze mit unterschiedlichen aktiven Räumen untersucht bis schließlich durch das Einfrieren inaktiver Orbitale eine Lösungsstrategie entwickelt wurde.
Die Topologie der Potentialfläche des elektronischen Grundzustandes ist sehr komplex, was durch die unterschiedlichen Minima und die Kopplung aller Freiheitsgrade hervorgerufen wird. Aus mehr als 240000 Datenpunkten konnte die Fläche konstruiert und mit einem künstlichen Neuronalen Netzwerk gefittet werden. Dieser Ansatz war sehr erfolgreich und führte nur zu kleinen Fehlern.
Der elektronisch angeregte Zustand wurde durch Entfernen eines Elektrons aus dem Wassermolekül realisiert. Dieser Ansatz steht im Einklang mit der experimentellen Beobachtung von Löchern, die das Adsorbat angreifen. Der Zustand wird bei molekularer Adsorption von stark repulsiven Kräften zwischen Adsorbat und Oberfläche dominiert. Die dissoziative Adsorption hingegen wird weiter begünstigt.
Die Potentialflächen wurden für quantendynamische Simulationen eingesetzt, bei denen das Adsorbat als Wellenpaket beschrieben wird. Die Photodesorption von Wasser wurde im Rahmen des jumping wave packet-Ansatzes studiert. Es zeigte sich, dass diese Reaktion ein Beispiel für den MGR-Mechanismus darstellt und zu schnell desorbierenden Molekülen führt.
Als zweite Reaktion wurde die Photodissoziation von Wasser untersucht. Dabei wurde die OH-Gruppe fixiert und nur die Bewegung eines Wasserstoffatoms simuliert. Die Dynamik wird durch eine kleine Energiebarriere in der Nähe des Franck-Condon-Punktes bestimmt. Diese verringert die Dissoziationswahrscheinlichkeit und führt zu ausgeprägten Isotopeneffekten.
Abstract (English)
In this thesis, the photochemistry of water on rutile is studied from first principles.
Surface photochemistry of water is important for a variety of applications and may lead to new routes for hydrogen production.
A cluster embedded in a finite point charge field is used as a model surface.
Systematic investigations ensured that parameters like cluster size, basis set and methods are sufficient for a good description of the water/rutile interaction.
It could be shown that a Ti9O18Mg714+ cluster, recently used for CO photodesorption, is large enough for water adsorption and dissociation, too.
On this cluster, a molecular and a dissociative adsorption form was found, the former being the most stable one.
Analyzing their geometries, five spatial degrees of freedom were identified as important for potential energy surfaces.
Construction of these surfaces is complicated due to the multi-reference character of the wave function in the course of the dissociation process.
Therefore, within the CASSCF approach a variety of active spaces was investigated. Finally, a two-step solution freezing the inactive orbitals paved the way for the potential energy surface.
The potential energy surface of the electronic ground state is very complicated because of different minima and a strong coupling between all degrees of freedom. The surface was constructed from more than 240000 data points and fitted by means of an artificial neural network. This approach proved to be very reliable giving only small errors and a good interpolation.
For the electronically excited state, one electron was removed from the water molecule. This is justified by the experimental observation of a hole attacking the adsorbate. This state is dominated by strong repulsive interactions between the adsorbate and the surface, as far as the molecular adsorption form is concerned. However, the dissociative form becomes even more favourite.
The potential energy surfaces were used for quantum dynamical simulations describing the adsorbate as a wave packet. The photodesorption of water in three dimensions was calculated within the jumping wave packet approach. The reaction was found to be a prototype of the MGR mechanism with very fast desorbing molecules.
The second reaction studied in this thesis is the photodissociation of water. Here, a simple system with a fixed \OH group was used to study the motion of hydrogen. The dynamics is controlled by a small energy barrier near the Franck--Condon point. This decreases the dissociation probability and leads to pronounced isotope effects.
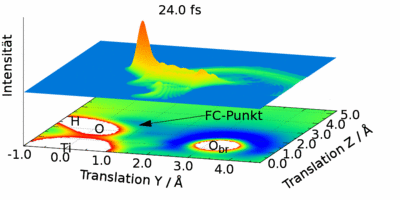
Quantendynamische Beschreibung eines Wasserstoffatoms als Wellenpaket.
Gezeigt ist die zeitliche Entwicklung des Betragsquadrats der Wellenfunktion bei einer fixierten OH-Gruppe auf der Rutil-Oberfläche. Es sind zusätzlich die Positionen einiger Atome und das Potential eingezeichnet.
Masterarbeit (2011)
Titel:
Ab initio-Studien zur Adsorption von CO auf C60
Bachelorarbeit (2008)
Titel:
Ab initio Rechnungen zu Struktur und Eigenschaften von Titandioxidoberflächen