Research
Contact
Prof. Dr. Christiane Richter-Landsberg
E-Mail: christiane.richter.landsberg@uol.de


![]()

![]()

![]()
Research
Research
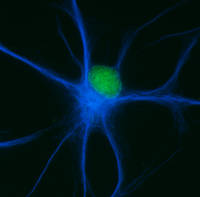
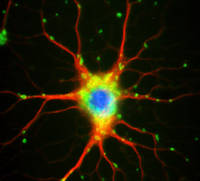
Nervous system functions are established by the coordinate interaction of nerve cells and glia, which might be impaired during aging and in neurodegenerative disorders. Our research is centered around oligodendrocytes, the myelin forming cells of the central nervous system, with a major focus on biochemical changes and molecular mechanisms regulating developmental processes, cell differentiation, cell survival, and cell death. At present the following major research areas are pursued:
- The functional roles of the oligodendroglia cytoskeleton during health and disease.
- The regulation of cell death and survival following cellular stress, and their underlying signal transduction pathways.
- Molecular mechanisms underlying the formation of glial cytoplasmic inclusions containing the microtubule associated protein tau and/or alpha-synuclein as observed in neurodegenerative diseases (tauopathies, synucleinopathies).
- The role of stress proteins and their induction in nerve cells and glia.
- Functional significance of posttranslational modifications of aggregation-prone proteins (ubiquitination, SUMOylation, phosphorylation)
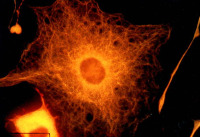
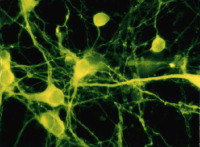
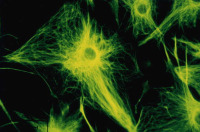
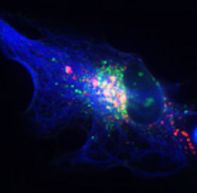
Mature oligodendrocytes in culture are characterized by their numerous cytoplasmic extensions and flat membranous sheets. These sheets contain an extensive cytoskeletal network of microtubules (MTs) and microfilaments. Microtubules participate in the elaboration and stabilization of the myelin-containing cellular processes, intracellular sorting processes, and provide the rails for organelle trafficking. These rails are especially important to guide mitochondria to the cytoplasmic extensions. In oligodendrocytes, the predominant microtubule-associated proteins (MAPs) are MAP2 and tau, which might be involved in the regulation and stabilization of the dynamic MT network.
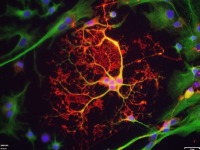
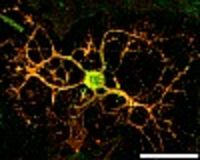
Alterations or disruption of the cytoskeleton and filamentous proteinaceous cell inclusions in nerve cells and glia are frequent hallmarks of neurodegenerative diseases and might cause cell death and degeneration. Glial changes occur in a variety of diseases. Glial cytoplasmic inclusions (GCIs) and inclusion bodies, containing abnormal microtubular structures, which stain positively for stress proteins, MAPs and ubiquitin, are found in oligodendrocytes of the affected brains.
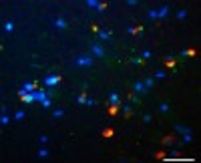
GCIs originating in oligodendrocytes are the histological hallmark of multiple system atrophy (MSA). MSA represents a progressive, sporadic neurological disorder with symptoms of Parkinsonism, ataxia and autonomic dysfunction. alpha-Synuclein is the major building block in GCIs but various reports demonstrate also the presence of tau. Emerging evidence indicates that tau and alpha-synuclein might overlap in the pathological features of inclusion body diseases, and possibly interact with and affect one another.
The microtubule associated protein tau in oligodendrocytes
We have shown that oligodendrocytes express all six isoforms of the microtubule associated protein tau. Oligodendrocytes are particularly sensitive to oxidative and proteasomal stress, causing upregulation of heat shock proteins and modulation of tau phosphorylation through the activation of protein phosphatase PP2A. Proteasomal inhibition causes the appearance of tau-positive fibrillary protein aggregates in oligodendrocytes. These also contain heat shock proteins and ubiquitin, and thus resemble glial cell inclusion bodies, as observed in frontotemporal dementias, e.g Progressive supranuclear palsy (PSP) and Corticobasal degeneration (CBD). Our recent work indicates that small heat shock proteins (HSP), mainly HSP25 and alphaB-crystallin associate with the cytoskeleton, contribute to survival responses and promote the solubility of tau in oligodendroglial cells. HSP32 might be a sensor of oxidative stress and is upregulated in oligodendrocytes during demyelinating processes.
Establishment of oligodendroglial cell culture model systems
Furthermore, we have developed an immortalized oligodendroglial cell line known as OLN-93. These we have engineered to express stably the longest human tau isoform with four microtubule binding repeats (OLN-t40) which is most prominent in tau inclusions in PSP, the shortest tau isoform with three MT-binding repeats (OLN-t44), or the longest tau isoform with the P301L mutation (OLN-P301L). This mutation has been linked to the formation of filamentous tau in oligodendrocytes, oligodendroglial apoptosis and the occurrence of 4-repeat hyperphosphorylated tau in transgenic mouse models. Using this cell line as a model system, we could demonstrate that proteasomal inhibition and tau hyperphosphorylation, exerted through okadaic acid, contribute to the formation of inclusion bodies. Also, proteasomal inhibition causes aggresome formation, which is accompanied by mitochondrial redistribution and cytoskeletal reorganization.
More recently we have engineered OLN cell lines coexpressing tau and alpha-synuclein which are used to study the molecular mechanisms underlying the aggregation process of these proteins.
Protein quality control systems during neurodegeneration and aging.
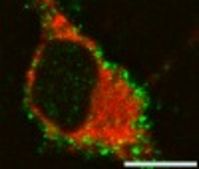
During neurodegenerative disease and aging the efficiency of the cellular quality control systems might be decreased or impaired. Aggregates may be formed when the levels of misfolded proteins overwhelm the degradative pathways, resulting in cellular toxicity and neurodegeneration. The two main cellular mechanisms for degrading abnormal or obsolete proteins include the ubiquitin-proteasome system (UPS) and autophagy, representing lysosome mediated degradative pathways. Short-lived nuclear and cytoplasmic proteins are marked by ubiquitin and are degraded predominantly by the proteasome, while long-lived structures and proteins are targeted to lysosomes by autophagy. Autophagy, specifically macroautophagy, plays a unique role in degrading and removing cellular organelles and protein aggregates that are too large to be degraded by the UPS. By autophagy, a cell can sequester regions of cytoplasm containing proteins or organelles forming a double-membrane bound vacuole, termed autophagosome. The autophagosome fuses with the lysosomes to form autolysosomes where their contents are then degraded by lysosomal acid hydrolases. Macroautophagy is particularly crucial in the aging brain, to protect cells from oxidatively damaged proteins and membranes, and aged or oxidatively damaged organelles. If the proteasomal system is unable to clear aggregates, autophagy may be the preferential route, and aggregate clearance can be enhanced by autophagy. Our recent data indicate that the autophagic pathway in astrocytes is activated by proteasomal inhibition and is involved in the removal of aggresomes. Furthermore, we have shown that the geldanamycin analogue, 17-AAG, not only causes the upregulation of heat shock proteins in oligodendroglial cells, but also is an effective inducer of the autophagic pathway by which alpha-synuclein can be removed. A major goal of our ongoing research is to investigate how autophagy is involved in the clearance of soluble and aggregated species of tau in oligodendroglial cells, and whether upregulation of autophagy in oligodendrocytes ameliorates tau aggregate formation and has cell survival promoting consequences.